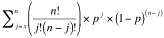CGD Help: GO Term Finder
Contents
The Gene Ontology (GO) project was established to provide a common
language to describe aspects of a gene product's biology. A gene
product's biology is represented by three independent structured,
controlled vocabularies: molecular function, biological process and
cellular component. For more information on GO, see SGD's GO Tutorial
or GO Help
pages, or see the GO consortium
home page.
To provide the most detailed information available, gene products are
annotated to the most granular GO term(s) possible. For example, if a
gene product is localized to the perinuclear space, it will be
annotated to that specific term only and not the parent term
nucleus. In this example the term perinuclear space is a child of nucleus.
However, for many purposes, such as analyzing the results of
microarray expression data, it is very useful to "calculate" on GO,
moving up the GO tree from the specific terms used to annotate the
genes in a list to find GO parent terms that the genes may have in
common. The GO Term Finder tool allows you to do this.
The GO Term Finder is described in detail in Boyle et al (2004).
The query page has several options as described below.
- Step 1: Choose Strain:
Select a strain from the pull-down menu.
- Step 2: Enter your gene(s):
You can either type the name of the genes in the input box or upload a
file that contains the gene names. Note that the program requires
more time to process a long list (greater than 100 genes) than a short
list.Either genetic names (CGD Standard Names, e.g., 'TUP1') or
systematic names (orf19 identifiers, e.g., 'orf19.6109') may be
used. The program handles alias and ambiguous names as follows:
- if a name is entered that is an alias name for one gene or
feature, the program will map the name to that gene. The CGD Standard
name or Systematic name will be displayed in the GO Term Finder
output.
- if a name is entered that is an alias name for gene A and also the
Standard or Systematic name for gene B, the program will map the name
to gene B. The CGD Standard name or Systematic name for gene B will be
displayed in the GO Term Finder output.
- if a name is entered that is an alias for more than one gene but not
a Standard or Systematic name for any genes, the program will present
a list of possible mappings. The user can determine which gene was
intended and go back to edit the input to use the CGD Standard or
Systematic name.
- Step 3: Choose your ontology:
Select one of the three (biological process, molecular function, or
cellular component) ontologies by checking the boxes. This tool is
designed to search only one of the three ontologies at a given time in
order to minimize the searching time.
- Click the Search button after Step 2 to search
using the default settings or go to Steps 3 and 4 to specify and customize
your background set and/or refine the annotations in your background
set.
- Step 4: Specify your background set.This is an optional step
that allows you to specify a background set of genes. The default
background set includes all the features/gene names in the database
that have at least one GO annotation.
You can also customize the background set of genes (default or
your specific set) by specifying feature type.
- Step 5: Refine the Annotations used for Calculations.
This is also an optional step and allows you to refine the annotations
to genes in your background set using three different criteria.
- Annotation Method refers to the methods used to generate
the experimental data on which the GO annotation is based, as well as
the curation method used to make the annotation. Manually
curated annotations are assigned for individual genes by curators
reading the scientific literature. High-throughput annotations
are assigned from published high-throughput or genome-wide
experiments; curators review the results as a whole but do not review
the annotations for individual genes. Computational annotations
are predicted by computational methods (e.g., sequence similarity
comparisons) and are not individually reviewed.
Note that the default set of annotations
used by GO Term Finder differs between CGD and
SGD. In CGD, all GO annotations (Manually curated, High-throughput,
and Computational) are used as the default set, while in SGD
Computational annotations are excluded from the default
set. Computational annotations are included for CGD because they
augment the GO annotation coverage of the genome, providing annotation
for many uncharacterized genes. In contrast, in SGD the greater extent
of characterization of S. cerevisiae genes means that computational
annotations are frequently redundant with or less specific than
experimentally-derived annotations, and dilute this higher-quality
set.
- Annotation
Source refers to the group that assigned the GO annotations:
either the Annotation Working Group (AWG; see Braun et al. (2005) A
human-curated annotation of the Candida albicans genome. PLoS
Genet. 2005 Jul;1(1):36-57), or CGD curators.
- Evidence Codes denote the
type of evidence that supports a GO annotation.
The results page displays, in both graphic and table form, the
significant shared GO terms (or parents of GO terms) used to describe
the set of genes entered on the previous page. In addition, the
results page displays all the criteria used to customize the
Background set and Annotations in the background set.
The graphic illustrates the relationships
among the GO terms used to directly or indirectly describe the genes
in your list. The color of each box indicates the p value score (see description
of the method below). Genes associated with the
GO terms are shown in gray boxes. Each GO term links to the CGD GO
term page, where you can view the GO structure around that term as
well as other genes associated with it. Each gene name links to its CGD
Locus Summary page.In some cases, the number of GO terms is too large to
display on a web page. When this occurs, the most significant terms
are shown. Regardless of the significant number of terms returned, an
option to download the complete set of results is always available.
To generate the graphics, the program utilizes CPAN's GraphViz perl wrapper module that uses
AT&T's
graphviz tool.
The table below the graph lists each significant GO term, the number
of times the GO term is used to annotate genes in the list (or
cluster) and the number of times that the term is used to annotate
genes in the background set. The default for the background set is
all the genes/features that have at least one GO
annotation in the database. The choice of background set is
configurable. Because the frequency of any given annotation within
the background set is compared against the frequency of the annotation
within the query set (input), the choice of background set affects the
significance of the results that are returned by the tool. Please
note that the specific background set of genes that was used in the
absence of any user-defined set (the default background set) has
changed over time. Prior to December 2007, the default background set
included all genes that have GO annotations in one or more of the
Biological Process, Molecular Function, or Cellular Component
ontologies. Between December 2007 and March 2008, the default
background set included only the genes that have a GO annotation in
the specific ontology that the user chose to query. As of March 2008,
the background set includes all of the genes in the genome, regardless
of whether or not they have GO annotations. In other words, the
stringency that is contributed by the background set has varied from
moderately stringent (before December 2007) to very stringent
(December 2007 to March 2008), and is now least stringent.
Additional columns list the p-value, the False Discovery Rate, and a
list of all the genes annotated, either directly or indirectly, to the
term. False Discovery Rate is an estimate of the percent chance that a
particular GO term that is shown as significant might
actually be a false positive. It represents the fraction of the
nodes with p-values as good or better than the node with this FDR
that would be expected to be false positives.
To determine the statistical significance of the association of a
particular GO term with a group of genes in the list, GO Term Finder
calculates the p-value: the probability or chance of seeing at least x
number of genes out of the total n genes in the list annotated to a
particular GO term, given the proportion of genes in the whole genome
that are annotated to that GO Term. That is, the GO terms shared by
the genes in the user's list are compared to the background
distribution of annotation. The closer the p-value is to zero, the
more significant the particular GO term associated with the group of
genes is (i.e. the less likely the observed annotation of the
particular GO term to a group of genes occurs by chance).
Results with a p-value less than 0.05 are color-coded on the graphical
display, and all results with a p-value of less than or equal to 0.1
are included in the results table. Please note that the same cutoff
has not always been used in generation of the results displays;
between December 2007 and March 2008 the cutoff for both displays was
increased in stringency from a p-value of less than or equal to 0.1 to
a p-value of less than or equal to 0.01, and subsequently relaxed.
The change in cutoff means that more results may be returned from any
given search, and (as always) the important task of judging which
results are truly significant under the circumstances is left to the
investigator.
A customizable
web implementation of the GO Term Finder tool that
allows the user to set a p-value cut-off (which uses the same
algorithm as the CGD's tool) is available at Princeton University.
Here are some important points to note when including results from
this tool in a publication.
- GO annotations are continuously updated at CGD. As a result,
one might not be able to reproduce a given set of results on a different
date. Mentioning the date when the analysis was done in the
publication can be useful.
- Mentioning details of the background set, including the number of
genes in
the background set and p-value cut-off used, can be useful.
Genes are directly associated with GO terms that are as granular as
possible. Because the GO terms have hierarchical relationships with
each other, genes are also considered to be indirectly associated with
all the parents of the granular terms to which they are directly
associated.
The tool looks for significant shared GO terms that are directly or
indirectly associated with the genes in the list. To determine
significance, the algorithm examines the group of genes to find GO
terms to which a high proportion of the genes are associated as compared
to the number of times that term is associated with other genes in the
genome. For example, when searching the process ontology, if all of
the genes in a group were associated with "DNA repair", this term
would be significant. However, since all genes in the genome (with GO
annotations) are indirectly associated with the top level term
"biological_process", it would not be significant if all the genes in
a group were associated with this very high level term.
Notes: This version of GO Term Finder uses a hypergeometric
distribution with Multiple Hypothesis Correction (i.e., Bonferroni
Correction) to calculate p-values. A stand-alone,
generic version of GO Term Finder that uses a hypergeometric
distribution, with Bonferroni Correction and False Discovery Rate, can be downloaded here.
Algorithm Details:
If G is the number of genes annotated to a term (either directly or
indirectly) and N is the total number of genes in the genome with GO
annotations (please see Results Table section above for details on
this number), then p, the probability of a randomly selected gene being
annotated to a particular GO term can be calculated as:
G
-
N
Given a list of n genes, in which x of them have been annotated to a
given GO term (directly or indirectly), the probability of having x
out of n annotations assigned to the same GO term by chance is defined
as the product of the number of permutations by which the annotations
can occur and the following equation:
px x (1-p)(n-x)
Within a list of n genes, there are multiple permutations by which x
of them may have this annotation. The number of permutations can be
calculated as:
n!
--------
x!(n-x)!
However, annotations to a particular term are low probability events
(p is small). Because of this, any list of genes having a particular
set of annotations is likely to have a low probability, but not
necessarily a significant one. Thus, instead of calculating the
probability of having x of n genes annotated to a term, a more
conservative approach, often used by statisticians, is taken to
calculate the probability of x or more of n genes being annotated to a
particular term. Since GO annotations are still incomplete (i.e. there
may be more than x genes annotated to a particular term), this is
appropriate. This is calculated as: